
Last Updated on 2023年3月16日
Hello, in this article, I’ll introduce the regulatory affairs (RA) regarding ethical drugs, classified as First-class marketing license in Japan. I think that the basic work contents are equally applicable to regulatory affairs other than ethical drugs. This time, I’ll explain roughly what kind of work dividing it into regulatory affairs positions on career change sites or job vacancy information, and also the relationship between each.
Regulatory affairs for ethical drugs / Pharmaceuticals in First-class marketing license
- Submit regulatory documents and apply to obtain the approval
- Consult with PMDA
- Coordinate not only clinical development but also other departments such as manufacturing and sales
Types of RA
RA’s positions (for ethical drugs / Pharmaceuticals in First-class marketing license) seem to be recruited for the following five categories. In particular, R&D (or just RA ), Post-marketing (lifecycle maintenance and compliance), and (R&D) CMC are typical three regulatory affair’s positions. However, depending on the size of the company and the items handled, positions may be added, or conversely, they may be combined into one position. I will mention about each position on the premise that the number and groupings differ depending on the company.
R&D RA
R&D RA works for the first and most important phase ‘apply – Obtain the approval’ before the life cycle of pharmaceuticals is created. R&D RA is recruited by companies handling new drugs, and in many cases, the following knowledge and experience are required in Japan.
- Plan (regulatory) timeline and strategy leading up to approval of the NDA in Japan
- Correspond and negotiate with PMDA (and Ministry of Health, Labor and Welfare)
- Prepare of regulatory dossier/documents (CTD, etc.)
- Keep up to the date with Japanese procedural requirements and regulation (Act on Securing Quality, Efficacy and Safety of Products Including Pharmaceuticals and Medical Devices, and related laws and regulations)
- English (to communicate with the foreign company or global colleagues)
The knowledge and experience listed above correspond to the advanced items in order from the top. In addition to planning strategies, important tasks in R&D RAs are dealing with and negotiating with PMDA (and the Ministry of Health, Labor and Welfare). Regarding correspondence and negotiations with PMDA (and the Ministry of Health, Labor and Welfare), R&D RAs will answer inquiries and explain and prove the propriety and/or compliance of quality, efficacy and safety. In a limited amount of time, R&D RAs are required to organize a huge amount of information and deal with the points and doubts of PMDA (and the Ministry of Health, Labor and Welfare) with flexible thinking. Especially with new drugs, unexpected situations can occur, so it is necessary to work with other departments to find the best possible response.
Post-marketing RA
Post-marketing RAs have a wider range of duties than other RA positions.
First, Post-marketing RAs mainly deal with products on the market after obtaining approval (In some companies, Post-marketing RAs seem to deal with also new drug approval applications). Specifically, Post-marketing RAs are in charge of change managing (minor change notification, partial change approval application, etc.) related to approval application forms that have been approved. There can be planned changes and sudden changes, so it is necessary not only to make a plan, but also to monitor in advance so as not to miss any sudden changes. Since Each pharmaceutical company is responsible for managing all pharmaceuticals on the market, the more products on the market, the more work. Since change management involves various changes related to the manufacturing site, such as manufacturing method/process changes and test method changes, Post-marketing RAs often work with CMC RA and Quality Control/Quality Assurance Department regarding GMP compliance inspection following Partial change approval application.
Secondly, Post-marketing RAs are responsible for managing business license, etc, such as marketing, manufacturing, and wholesale distribution. The licenses are required to be updated regularly and the quantity is large. I don’t think it’s unlikely, but if the licenses are not updated, the business will be forbidden, so managing business licenses requires a big responsibility. In addition, Post-marketing RAs may be required to be responsible for responding to GMP compliance inspections as a staff of the company’s Quality Assurance Department.
The third is regulatory reveiwing and support for advertising and promotion. Those activities and materials must be ensured with regulatory compliance.
In addition to the above wide range of duties, they may also be responsible for creating labeling and package inserts. Therefore, I think that the scope of Post-marketing RAs varies greatly depending on the company.
CMC RA
Compared to R&D RAs and Post marketing RAs mentioned above, CMC RAs are more closely related to manufacturing and quality. CMC RAs may belong to a department related to CMC, a factory or a research center instead of the RA department. CMC stands for “Chemistry,” “Manufacturing,” and “Control.” CMC RAs handle Manufacturing process of active pharmaceutical ingredients and products, manufacturing procedure, specification and testing method, stability, physicochemical characterization, batch analysis, then they integrate to CTD and the application form as data or documents. Many active pharmaceutical ingredients are manufactured overseas, so it is necessary to translate documents and reports, and to confirm and respond to whether the documents and data meet the requirements for Japanese regulations. Since CMC RAs are related to manufacturing, CMC RA’s tasks include PCA, MCN, GMP compliance inspection, Import/export of drug substances and products, and etc, in some cases.
I’m not familiar with CMC RAs well. Through looking at the CMC RA’s recruitment, there are cases where RA experience (not CMC) is written in the qualification for CMC RA. In some pharmaceutical companies, (especially Post-marketing) RA prepare for submitting data and answering to queries from PMDA (and the Ministry of Health, Labor and Welfare) regarding CMC. And (R&D or Post-marketing) RAs have opportunities to learn about the work of CMC through their work. Therefore, even if RAs do not know details about CMC RA at first, I think (R&D or Post-marketing) RAs can gain more or less experience in CMC through tasks of (R&D or Post-marketing) RAs.
Electronic submission / Submission (management)
The position regarding electronic submission management in new drugs is a relatively new position in Japan. In 2003 , Transferring electronic of CTD (Common Technical Documents) for NDI start to be discussed, and PMDA started accepting eCTD since Apr 1, 2005, which is an electronic medium (CD, DVD, etc.) in Japan. From October 1, 2016, it became possible to accept applications to the PMDA through communication from a portal site on the web called Gateway, after repeated improvements and discussions with industries. Until March 31, 2020, as a grace period, PMDA accepted not only electronic media applications, but also direct delivery and mailing of paper media, but from April 1, 2020, only eCTD gateway applications will be accepted in principle. On the other hand, applying to get MA of generic drugs are still mainly submitted by paper CTD.
Electronic submission / Submission management is required to create and convert the CTD to electronic media as eCTD according to the appropriate standards for electronic submission, and to organize it into an appropriate file so that errors do not occur at the time of submission. Not only the eCTD at the time of submission, but also electronic data submission required by PMDA, receipt of inquiries after the submission, responses to inquiries, and resubmission when necessary, will be handled through the gateway.
Electronic submission / Submission management vacancies are available at large companies selling new drugs. This is because it is more efficient for large companies to separate the function/position to deal a lot of pipeline and items requiring electronic submission. Since the amount of work is not large enough for small and medium-sized companies to specialize in, I think that (R&D or Post-market) RA will be responsible for electronic submission / submission management. There are many CROs who are proficient in the field of Electronic submission, and I believe that there are many pharmaceutical companies that rely on the help of CROs. Coordination and management with CRO will also be part of electronic submission / submission management. As a result, it seems that they have set up a separate department from the existing RA department to differentiate themselves.
RA Document and Clinical trial protocol notice staff
This position is described as above because I do not know the distinguishable position name. This is a position (in a broad sense, as a RA) recruited by CRO. As far as I can confirm, I don’t think that the above job offers have been posted by pharmaceutical and medical device companies. Duties include submission of clinical trial notice, creation of informed consent document, support for creation of clinical trial protocol, support for creation of investigator’s brochure, and QC / review of CTD and PMDA consultation materials. There are many supports and QC work, and it is often written as welcoming inexperienced in recruitment.
I think this position is closer to development than RA. First of all, it depends on the company whether the clinical trial notice is submitted by the (clinical) development department or the RA department. Except for the initial submission of the clinical trial notice, most of the information related to medical institutions, support institutions, and investigators in clinical trials is updated, so I do not think that RA necessarily need to respond. it is actually and occasionally not dealt with by RA but clinical development. Next, regarding informed consent document, clinical trial protocol, and investigator’s brochure, judging from the duties of the recruitment, it is considered to be a clinical development work rather than a RA work. On the other hand, PMDA consultation materials and CTD can be said to be RA’s work. However, I’m not sure if the work related to PMDA consultation materials and CTD, which can be said to be RA’s work, is limited to QC and reviews, and whether it will lead to a career path as a RA. As a result, I have the impression that most of the work involved is clinical development rather than RA.
If I have a chance, I would like to gather information of the RA’s work in CRO.
Summary
I have introduced the RA business by dividing it into 5 positions as above. I don’t know yet everything about RA, so the contents of the degree that I saw and heard are also included. Although it is simple, the figure below summarizes which stages each RA is mainly responsible for from the development of ethical drugs to the market in Japan.
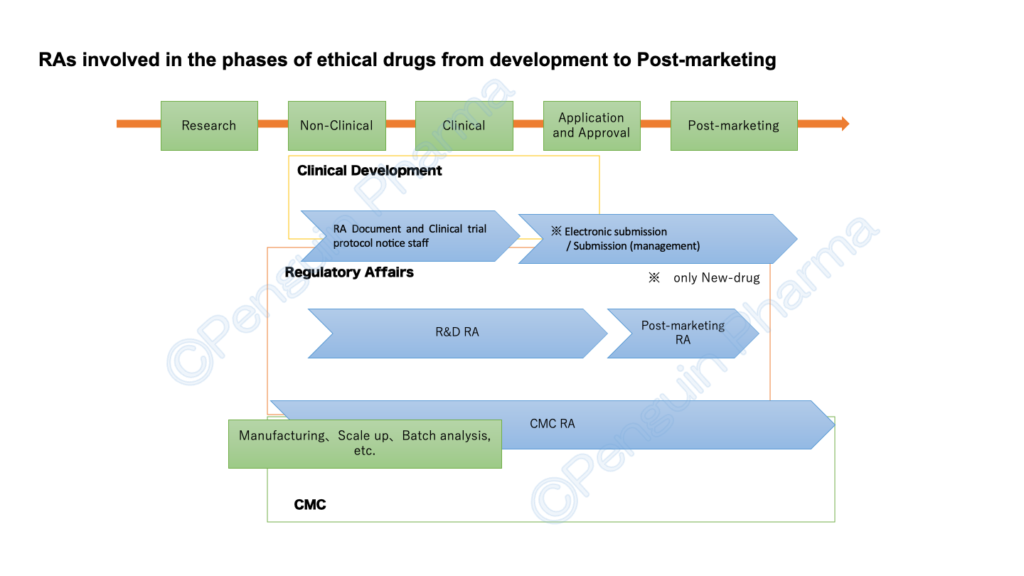
Although it is rough, I would appreciate it if I could correctly convey the relationship between RA and the work. As mentioned at the beginning, departments and organizational structures related to RA have different names and categories depending on the company. In the future, I would like to introduce more detailed work related to RA.
Although the relationship between RA and work is complex, your explanation helps clarify its nuances. It’s interesting how departments and structures vary across companies, reflecting diverse approaches. Introducing more detailed RA-related work in the future would be valuable for deeper understanding. Could you elaborate on how RA impacts workflow efficiency specifically?
Understanding the relationship between RA and the work seems complex due to varying department names and structures across companies. It’s great that the author aims to clarify this connection further. I’m curious to see how the detailed work related to RA will be introduced in the future. Could you provide examples of how RA impacts different organizational structures?
The relationship between RA and the work seems complex and not widely understood. It’s interesting how different companies categorize RA within their organizational structures. I wonder if the variations in naming and categorization create confusion or inefficiencies. It would be helpful to know more about the specific tasks and responsibilities involved in RA work. Are there common challenges that professionals in this field face across industries? Introducing more detailed work related to RA could provide clarity and improve collaboration. What do you think is the most crucial aspect of RA that needs to be better communicated or standardized?
Interesting perspective on the relationship between RA and the work! It’s true that RA’s role can vary so much across companies, which makes it a bit tricky to define universally. I appreciate your effort to clarify this, even if it feels rough at the moment. It would be great to see more detailed examples of RA-related work in the future—it could really help others understand its importance. Do you think the lack of standardization in RA roles across industries creates challenges for professionals in this field? I’m curious to hear your thoughts on how RA could be better integrated into organizational structures. What’s one thing you wish more people understood about RA?
The relationship between RA and the work seems complex but intriguing. It’s interesting how different companies categorize RA-related departments so differently. I wonder if this variability affects the efficiency or clarity of RA’s role. Your intention to delve deeper into RA-related work in the future is commendable—it could bring much-needed clarity. Do you think standardizing RA’s role across industries would be beneficial? I’d love to hear your thoughts on how RA could evolve to better align with organizational goals. What challenges do you foresee in introducing more detailed RA work?
Although RA’s role varies across companies, it’s crucial to understand its core relationship with the work. I found your explanation insightful but would love deeper examples of how RA impacts day-to-day operations. Could you elaborate on specific challenges RA faces in different organizational structures? I also wonder if there are any common misconceptions about RA that you’ve encountered. Your future introductions to RA-related work sound promising—will they include case studies or real-world applications? Lastly, do you think RA’s role will evolve significantly in the next few years? Looking forward to your thoughts!
The relationship between RA and the work seems complex but intriguing. It’s interesting how different companies categorize RA departments so differently. I wonder if this variability affects the efficiency or clarity of RA-related tasks. Your intention to introduce more detailed work on RA is promising—will this include case studies or specific examples? I’d love to hear more about the challenges you’ve faced in conveying this relationship. Do you think standardizing RA terminology across industries could be beneficial? Looking forward to your insights!
Understanding the relationship between RA and work seems crucial for clarity in organizational structures. I find it interesting how different companies have varied names and categories for departments related to RA. This makes me wonder if there’s a standard framework that could simplify this across industries. Your intention to delve deeper into RA-related work is commendable. I think it would be beneficial to explore case studies or examples to illustrate these differences more vividly. Do you believe there’s a chance to standardize these categories in the future? I’m curious to hear your thoughts on whether this variability actually benefits companies or if it complicates collaboration.
Контген: Про тол ПОlмза but rough stuff, I would be happy if I could rightly put on the relationship between RA and the work. As said earlier, RA associated departments and organizational structures vary in name and size depending on the company. In the future, I would like to introduce more detailed works related to RA.
This text provides a good starting point for understanding the relationship between RA and work, but it feels a bit general. The mention of different departments and organizational structures is interesting—why do these variations exist? Are there specific industries where RA is more prominently structured? I’d love to see examples of how RA influences daily tasks or outcomes in practice. The promise of introducing more detailed work in the future is exciting—will this include case studies or real-world applications? Overall, it’s a solid foundation, but I’m curious to see how it evolves. Would you say RA is more about compliance, strategy, or both? Looking forward to your next insights!
This is an interesting perspective on RA and its relationship with work. I appreciate the effort to clarify the different names and categories across companies. It’s true that RA can be complex, and breaking it down further would be helpful. I wonder if there are specific challenges in standardizing these structures across industries. Could you share examples of how RA impacts day-to-day operations in different sectors? I’d love to hear more about the detailed work you plan to introduce. What do you think is the most misunderstood aspect of RA? Looking forward to your insights!
It’s interesting how RA’s role varies so much across companies—it really highlights the complexity of the field. I think it’s great that you’re aiming to clarify these relationships, as it can be confusing for many. The idea of introducing more detailed work on RA sounds promising and could be very helpful. Do you think the differences in naming and categorization hinder collaboration between companies? I’d love to hear more about how you plan to address these variations. What specific aspects of RA work do you think need the most attention? This seems like a topic that could spark a lot of discussion—what’s your take on it?
This is an interesting perspective on the relationship between RA and work. It’s true that RA’s role can vary widely across companies, which makes it a bit complex to define universally. I appreciate your effort to clarify this relationship, as it’s often overlooked in discussions. It would be great to see more detailed examples of RA-related work in the future—it could help others understand its importance better. Do you think the lack of standardization in RA roles across companies creates challenges for professionals in this field? I’m curious to hear your thoughts on how RA could be more effectively integrated into organizational structures. What steps do you think companies should take to make RA roles more consistent and impactful?
The relationship between RA and the work seems complex and varies across companies, which makes it quite intriguing. It’s interesting how departments and organizational structures differ so much—it must be challenging to navigate. I’d love to hear more about the specific challenges you’ve faced in this area. Could you elaborate on how RA impacts day-to-day operations in different companies? Also, what kind of detailed work related to RA are you planning to introduce? It sounds like there’s a lot of potential for deeper insights here. Do you think standardizing RA-related structures across industries would be beneficial, or does the diversity add value? Looking forward to hearing your thoughts!
This is an interesting take on the relationship between RA and work. It’s true that RA can vary so much across companies, which makes it a bit confusing at times. I appreciate the effort to clarify this, but I wonder if there’s a way to make it even more straightforward. Do you think there’s a universal framework that could apply to RA across industries? Also, I’m curious about the specific challenges you’ve faced in defining RA in your work. Looking forward to hearing more about the detailed work you plan to introduce—it sounds promising! What’s the most surprising thing you’ve learned about RA so far?
This is an interesting perspective on RA and its relationship with work. It’s true that RA can vary so much across companies, which makes it a bit confusing to understand its exact role. I appreciate the effort to clarify this, but I’m curious—how do you think RA could be standardized across industries? Also, what specific aspects of RA work do you plan to introduce in the future? It would be great to hear more about your vision for RA’s role in organizational structures. Do you think RA is often underestimated in its importance? I’d love to hear your thoughts on how RA can be better integrated into company workflows. What challenges do you foresee in achieving this?
The relationship between RA and the work seems complex, especially with the varying departmental structures across companies. It’s interesting how RA’s role can differ so much depending on the organization. I appreciate your effort to clarify this connection, as it’s not always easy to explain. Introducing more detailed work related to RA in the future sounds promising—it could provide much-needed clarity. Do you think the lack of standardization in RA’s role across companies creates challenges? I’d love to hear your thoughts on how RA could be better integrated into organizational frameworks. What specific aspects of RA’s work do you think are most misunderstood?
This is a very insightful piece on RA and its varying roles across different organizations. It’s interesting how RA seems adaptable yet complex based on company structures. I’d love to know more about the specific challenges faced in defining RA’s role—do you think standardization is possible, or is flexibility its strength? The mention of future plans about detailed RA work is intriguing, but I wonder if there’s a priority area you’re focusing on first? Also, how do you see RA evolving in the next few years? Looking forward to your thoughts!
By the way, we’ve integrated libersave into our regional voucher system. It’s amazing how easily it brings together various providers on one platform!