
Last Updated on 2023年3月15日
こんにちは、ペンギンです。今回は、医療用医薬品の薬事の業務について紹介したいと思います。医療用医薬品以外の薬事においても基本的な業務内容は等しく当てはまるのではないかと考えます。今回は転職サイトや求人で募集されている薬事ポジションに分けてどのような業務かについてざっくりと説明するとともにそれぞれの関係性についても触れていきたいと思います。
医療用医薬品の薬事業務について
薬事の業務とは何かと考えたときに一概に答えることはできませんが、私が臨床開発職に従事していたときには薬事の業務について以下のようなイメージを持っていました。
申請に関わる業務
PMDAとの相談に関わる業務
臨床開発だけでなく製造や販売の他部署をまとめる業務
実際に薬事職として従事してみると、もともとのイメージ通りであったこともあればイメージ通りではない部分もありました。特に面白いポイントは企業によって薬事の業務範囲が異なるというところです。薬事の業務はMRのように認定を受ける必要もなければ薬剤師の資格の取得が必須でもありません。従って、薬事の業務に明確な線引きはないと考えています。例えば、大手企業であれば品目も多く膨大な業務を細分化する必要がありポジションが増えることはみなさま納得できると思います。そして、中小企業では品目が少ない一方で一人一人の薬事担当者が幅広く業務を担うことが予想されます。一方で、企業規模とは別に初回申請や戦略をどの部署が請け負うかという違いもあります。例えば企業によっては(臨床)開発の部署で初回申請や戦略を担うそうです。従って薬事に関する手順を習得し、対応できる能力を持つ担当者がいずれかの部署にいれば薬事部である必要はないと私は考えています。実際には、特定の人に業務が過度に集中してしまいますので推奨されませんが…
薬事(医療用医薬品)の種類
医療用医薬品の薬事ポジションは以下の5つについて分かれて募集されていると見受けられます。特に開発薬事、薬制薬事、CMC薬事の3つは代表的な薬事ポジションだと思っています。ただし企業規模や取り扱い品目によってはポジションが加わったり、逆に一つのポジションにまとめられていたりします。企業によって人数も括りも異なることを前提に各ポジションについて述べていきます。
開発薬事
開発薬事は医薬品のライフサイクルが生み出されるまでの最初にして最重要な”申請〜承認”段階を担う薬事となります。開発薬事の求人は新医薬品を扱う企業で募集され、多くの場合、以下の知識や経験が求められます。
- 承認にいたるまでの(薬事)戦略の策定
- PMDA(や厚労省)との対応、交渉
- 申請書類の作成(CTDなど)
- 薬機法や薬事関連法令などの情報の収集
- 英語力
どの求人でも比較的同様の内容が書かれていますが、経験や知識量は各社の開発を進める品目あるいは領域に対して高いレベルで求められ、責任を持って対応できるようになるまで経験を積み上げる道のりは長いです。また、上記以外で求められる業務について例を挙げますと添付文書案の作成や外国製造業者認定申請、薬価基準収載手続きまで担う場合もあります。(大企業では特に)各役割に担当者がそれぞれにいる場合もありますが申請書類をまとめていく過程で薬事がレビューや取りまとめをすることが多く、また全体を通して薬機法や薬事関連法令を遵守しているか監視する必要があります。結果として安全性、薬物動態、海外での使用状況を含めて広く相当の知識が求められるとともに他部署との連携が強く求められます。
上記に列記した知識や経験は上から順に高度な項目にあたると考えます。特に戦略の策定とともに開発薬事で重要な業務が厚労省やPMDAとの対応、交渉となります。厚労省やPMDAとの対応・交渉については照会(問い合わせ)に対して回答して品質、有効性や安全性に問題ないことを説明・証明していきます。時間が限られる中で膨大な情報を整理して、柔軟な思考で厚労省やPMDAの指摘や疑義事項に対処が求められます。特に新医薬品では想定外の事態も起こり得ますので他部署とも連携して最善の対応を導くことが必要です。
薬制薬事
薬制薬事は他の薬事職と比べると業務の幅が広いです。
一つ目として主に承認取得した後に販売する品目を扱います(企業によっては薬事申請業務に関わるせいか新薬承認申請の業務も担っているようです)。具体的には承認を得た承認申請書に関わる変更管理(軽微変更届や一部変更承認申請など)を担います。計画的な変更もあれば突発的な変更も起こり得ますので計画を立てるだけでなく、急な変更を事前に見逃さないように監視する必要があります。製造販売元である医薬品全て管理する責任があるため市場に多く出回っている品目が多いほど対応する業務も多くなります。変更管理の業務は製法・工程変更,試験法変更などの製造所に関する変更が多いことから一部変更承認申請に伴ってGMP 適合性調査に関係する機会もあり、品質管理/品質保証部門やCMC薬事との連携頻度も多いです。
二つ目として製造販売業、製造業、卸売販売業などの業態管理を担います。定期的に更新が必要であり取り扱う数量も多いです。まずないとは思いますが対応を疎かにすれば業務停止になりますので責任も大きいです。また品質保証部門としてGMP適合性調査対応を担う場合もあるようです。
三つ目は、プロモーション活動の薬事的なレビュー及び支援です。薬事規制に沿う必要があります。
以上の幅広い業務に加えて添付文書の作成を担う場合もあり、その業務の幅広さから会社によって薬制の範囲が大きく異なるのではないかと思います。
CMC薬事
CMC薬事は上記の開発薬事と薬制薬事と比べて製造や品質に強く係る薬事となります。薬事と名はつきますが薬事部ではなくCMCに関する部署であったり工場や研究センターに所属している場合もあります。CMC とは「Chemistry(化学)」「Manufacturing(製造)」「Control(管理)」のそれぞれの頭文字を取った略で、原薬及び製剤の製造方法、製造工程、規格及び試験法、安定性および物理化学的特性から原薬の製造スケールアップ時のロット分析などを取りまとめ、最終的にCTD や申請書に書類・データとしてまとめます。特に原薬については海外で製造されている場合も多いため、資料や報告書の翻訳や、資料やデータが日本での申請・規制用件を満たすかどうか確認・対応する必要があります。CMC薬事は製造に関わるため、承認取得後も継続してフォローしていくことが必要になりますので時に一部変更申請書、軽微変更届やGMP適合性調査そして原薬や製剤の輸出入に関する業務なども任されることと思います。
CMC薬事については私もまだ全容を把握できていないですが、CMC薬事の求人を見ると(CMCではない)薬事経験者が条件として認められる場合もあります。企業によっては(特に薬制)薬事が厚労省やPMDAに資料提出・照会回答の対応においてCMC領域を担う場合があり、あるいは(開発あるいは薬制)薬事が業務を通してCMCの業務を知り得る機会があります。従って、最初はCMC薬事業務について知り得ない場合でも、CMC薬事について経験を大なり小なりは積むことができるという印象を私は持っています。
電子申請/申請マネジメント(新医薬品のみ)
新医薬品における電子申請/申請マネジメントと呼ばれる職種は比較的新しくできたポジションになります。日本では新医薬品の承認申請について2003年(平成15年)にCTD(コモン・テクニカル・ドキュメント)の電子化が標題となり、2005年(平成17年)4月1日より電子化コモン・テクニカル・ドキュメント(通称、eCTD)という電子媒体(CDやDVD等)による申請資料のPMDA受付が開始されました。それから公民ともに改良や協議を重ねゲートウェイというWEB上のポータルサイトから通信によってPMDAへの申請受付が2016年(平成28年)10月1日より可能となりました。2020年3月31日までは猶予期間として電子媒体による申請以外にも紙媒体による直接搬入・郵送も受付しておりましたが、2020年4月1日から原則としてeCTDゲートウェイ申請のみが受付されることととなりました。一方で、後発医薬品の申請資料は現在も紙のCTDによる提出が主流となっています。
電子申請/申請マネジメントは、電子申請にあたってCTDを適切な規格でeCTDとして電子媒体に作成変換し、申請時にエラーが出ないように適切なファイルにまとめることが必要になります。申請時のeCTDだけではなくPMDAが求める電子データの提出や申請以降の照会事項受領および回答提出、および必要時期の再提出についてもゲートウェイを通しますので、申請時・提出時のとりまとめ業務を担うこととなります。
電子申請/申請マネジメントの求人は、新医薬品を扱う大企業で募集されています。なぜなら大企業ではパイプライン、すなわち申請品目が多く電子申請業務を専業として分けたほうが効率が良いからです。中小企業では専業にできるほどの業務量ではないため恐らく(開発あるいは薬制)薬事が電子申請/申請マネジメントについて引き受けていることになると思います。電子申請の領域に長けたCROも多く、CROの手を借りている製薬企業も多いと思います。CROとの連携やマネジメントも電子申請/申請マネジメントの業務にあたるでしょう。結果として、今までの薬事部門とは別部門を立ち上げて差別化していると見受けられます。
薬事資料作成/治験届担当
こちらのポジションは、区別できるポジション名を知らないため上記のように記載しました。CROの求人において(広い意味で薬事職として)募集されているポジションです。確認するかぎり医薬品、医療機器メーカーで上記求人が出ていたことはなかったと思います。業務内容としては治験届の提出、同意説明文書の作成、治験実施計画書の作成サポート、治験薬概要書の作成サポート、治験相談資料やCTDのQCやレビューなどが挙げられます。サポートやQC業務が多く求人にも未経験可能と銘打っている場合が多いです。
こちらのポジションについては薬事よりかは開発に近いポジションと考えています。まず治験届は企業によって、(臨床)開発の部署が提出するか薬事の部署が提出するか異なります。治験届の初回提出時を除けば臨床試験における医療機関や支援機関、そして治験実施医師に関する情報の更新がほとんどのため必ずしも薬事が対応必要とは思いませんし、実際に薬事ではない(臨床)開発の部署で対応されている企業もあると思います。次に同意説明文書、治験実施計画書および治験薬概要書に関しても求人の職務から察するに薬事としての業務より開発としての業務と考えられます。一方で、治験相談資料やCTDであれば薬事に関する業務と言えると思います。しかし、薬事の領分といえる治験相談資料やCTDに関する業務がQCやレビューだけになると薬事としてのキャリアパスにつながるか心許ないです。結果として業務内容は薬事よりかは開発寄りの業務が多いという印象を私は持っています。
機会があればCROにおける薬事業務についても調べていきたいと思います。
まとめ
以上、薬事の業務について5つの求人のポジションに分けて紹介いたしました。私自身が、とても全てを知っているほどではありませんので見聞きした程度の内容も含まれています。簡単にではありますが医療用医薬品の開発から市場に出るまでの間で各薬事が主にどの段階を担うか下図にまとめてみました。
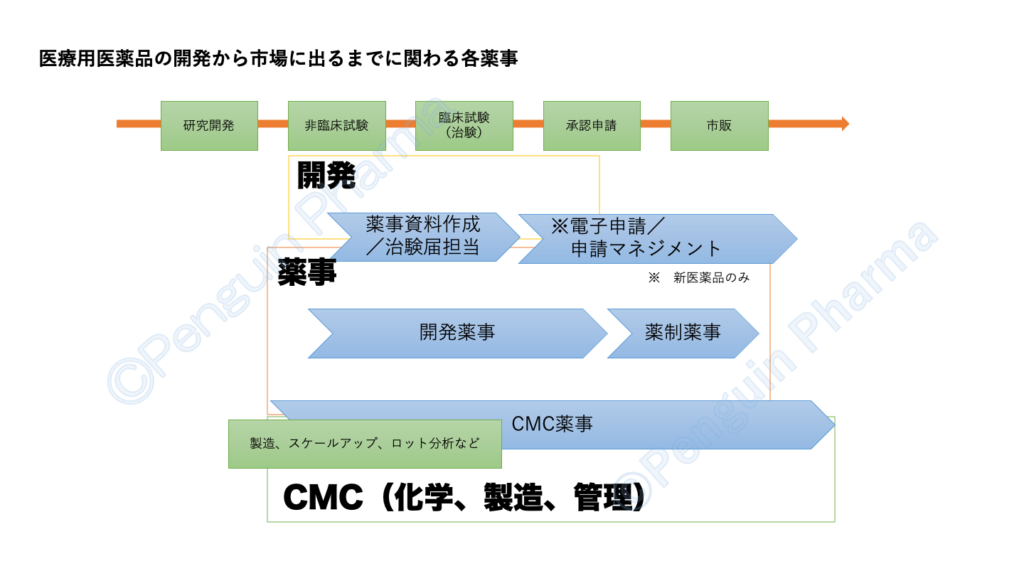
大まかではございますが薬事の業務と関係性を正しく伝えることができましたら幸いです。冒頭でも述べましたように薬事に関わる部署や組織体制は企業によって名称も括りも異なります。今後、薬事に関する細かい業務についても改めて勉強がてら紹介していきたいです。
Thank you for the comprehensive and insightful content on this website. Your commitment to educating and empowering others is truly admirable. The articles are not only informative but also engaging, making learning a truly enjoyable experience. Thank you for sharing such valuable information and contributing to our growth. Goodbye! ID : CMT-QMIYUBKP4B5Y1ULPC5